Introduction:
The broad goal of our research program is to understand the molecular mechanisms that control gene expression in early development. More specifically, we use genetic and molecular approaches to study transcriptional regulation in the early Drosophila embryo. We study both the transcription factors that regulate genes and the DNA sequences they interact with – enhancers and promoters.
Current Projects
Zelda’s role in genome activation
Arguably the most important transcriptional event in development across all metazoans is genome activation, the process by which the embryo transitions from a transcriptionally silent state to an active one. Our lab discovered the critical gene that coordinates Drosophila genome activation, the pioneer transcription factor Zelda. Zelda has been implicated in multiple processes that control the 3D organization of the genome, both in terms of the distribution of transcription factors and localization of genes and regulatory elements. This project is directed towards following up on how these observed spatial features that Zelda controls relate to its ability to drive transcription in the early embryo.
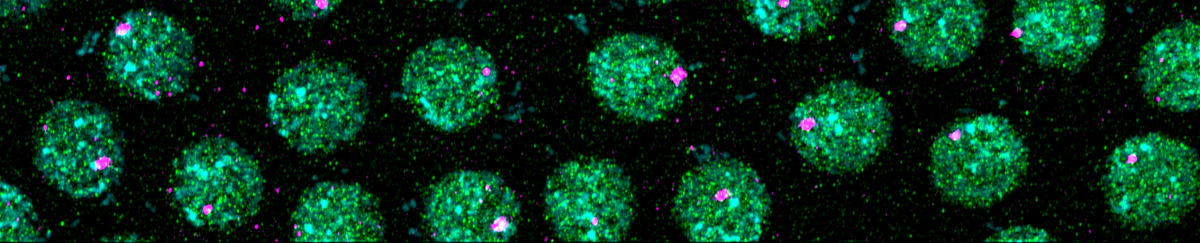
Drosophila is the ideal model system to study genome activation because of its extensively studied gene regulatory networks in early development, and the wealth of available tools that have been developed over decades of research. In particular, the Drosophila embryo lends itself extremely well to imaging assays. Here, we can visualize and quantify the process of genome activation through antibody staining of the major subunit of RNA polymerase II. Foci in the staining appear when active transcription takes place, and the number of foci is directly related to the number of genes that become active during genome activation.
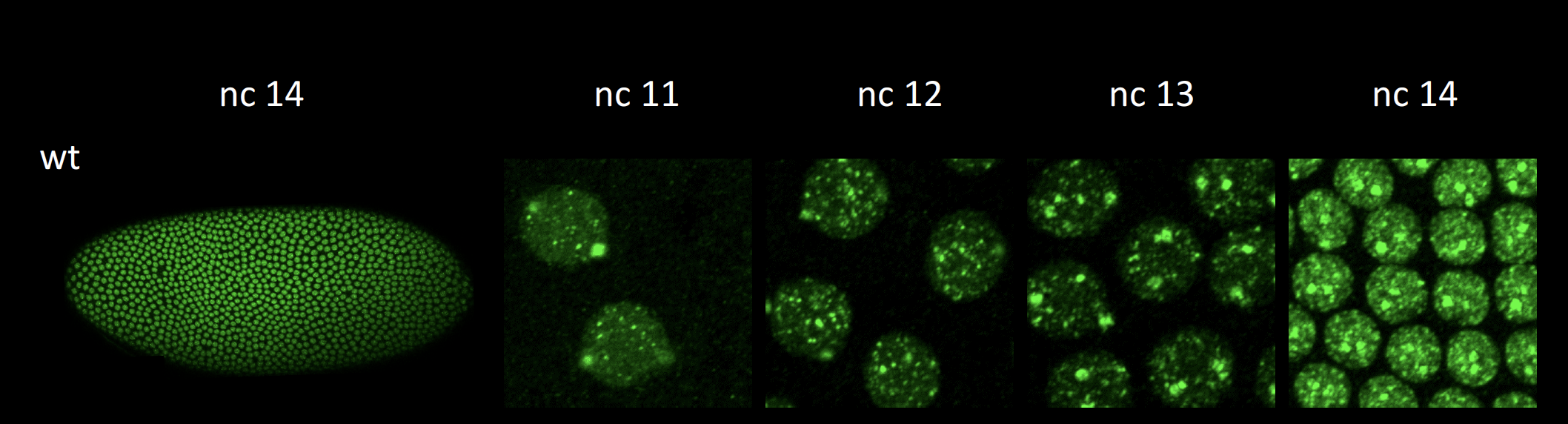
We can also visualize the location of single genes, and quantify their output in a single cell manner by examining the nascent transcripts in each nucleus. This is done through fluorescent in situ hybridization (FISH), which uses antisense RNA probes for specific genes. Nascent transcripts create large foci of staining, which co-localizes with Pol II staining.
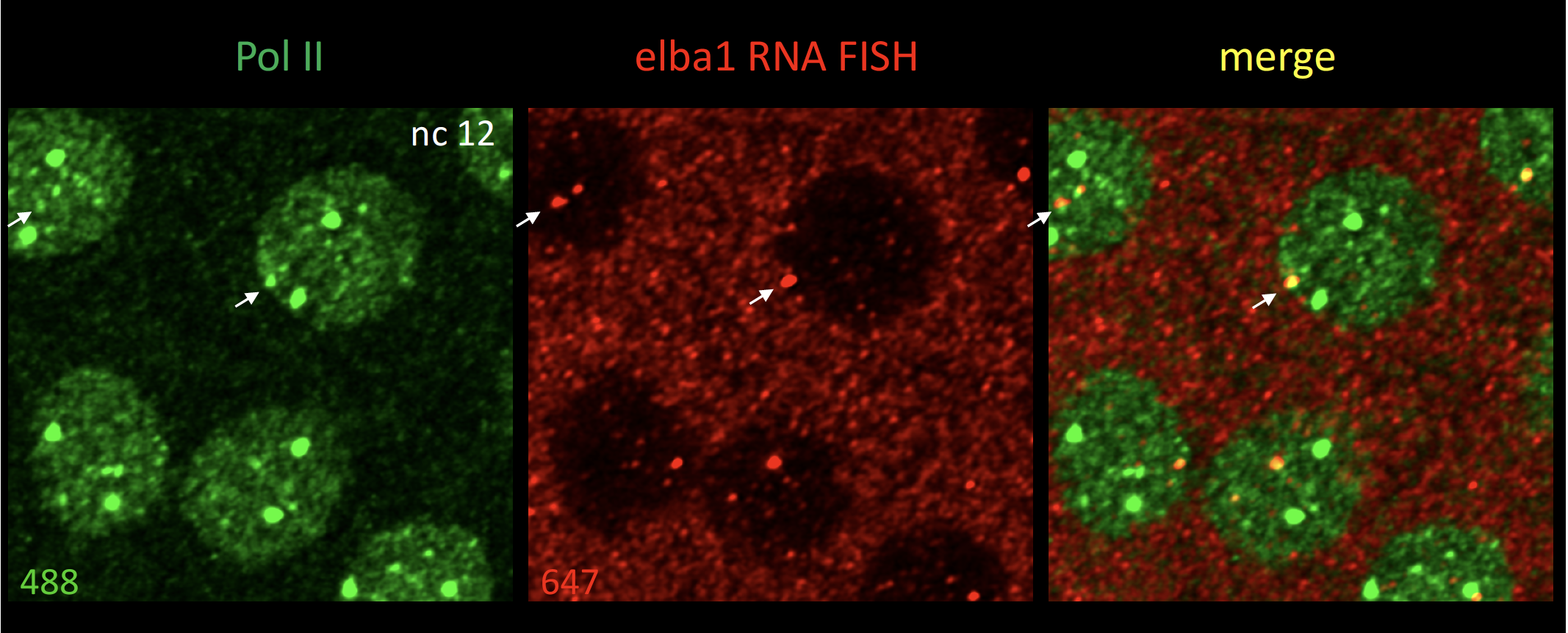
We are currently testing the idea of transcriptional hubs comprise of several genes, which may lead to more coordinated or robust transcription. Preliminary results show that genes located very close to each other, for example, within 5 kb, exhibit a single Pol II focus by confocal imaging, indicating that the two genes share an aggregation of Pol II.
Zelda as a Pioneer Factor
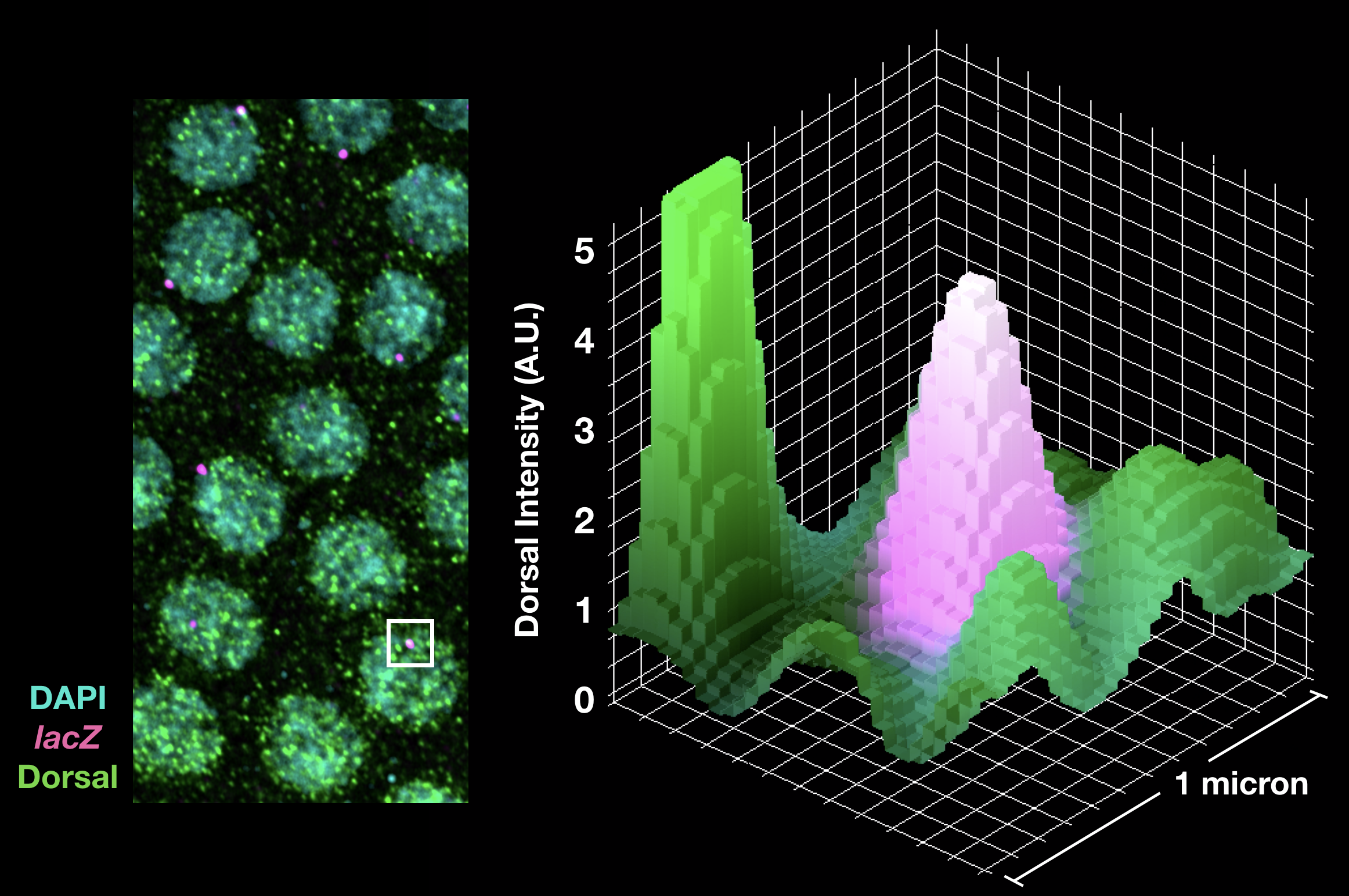
Pioneer factors represent a unique subset of transcription factors that can bind to nucleosomes and convert “closed” chromatin to “open” chromatin. Our latest publication investigated the transcriptional affects Zelda’s pioneering activity (Yamada and Whitney et al 2019). We found that Zelda bound enhancers show increased aggregation of transcriptional activators around the site of transcription, which led to more robust activation across a transcription factor morphogen gradient.
Enhancer Interactions
The coordination of gene expression in early development is critical to establish the proper body plan of all multicellular organisms. Transcription of these genes are controlled principally by non-coding DNA elements known as enhancers. Many genes rely on multiple enhancers to activate expression in overlapping domains that confer developmental robustness. So-called “shadow enhancers” are thought to buffer gene expression from environmental and genetic perturbations allowing organisms to reliably produce precise transcriptional output. This project is focused on dissecting the molecular mechanisms that integrate input from multiple enhancers.
To do this, we use high resolution imaging to view transcription in real-time with the MS2 imaging system in live Drosophila embryos. This technique allows us to infer transcriptional kinetics on enhancer deletion backgrounds. By understanding how enhancers operate in isolation and synergistically, we can learn the principles that govern enhancer behavior in complex multi-enhancer systems. This allows us to better understand not only how patterning signals are reliably interpreted in the early Drosophila embryo, but also to create a more complete theory of gene expression that applies across multiple organisms.
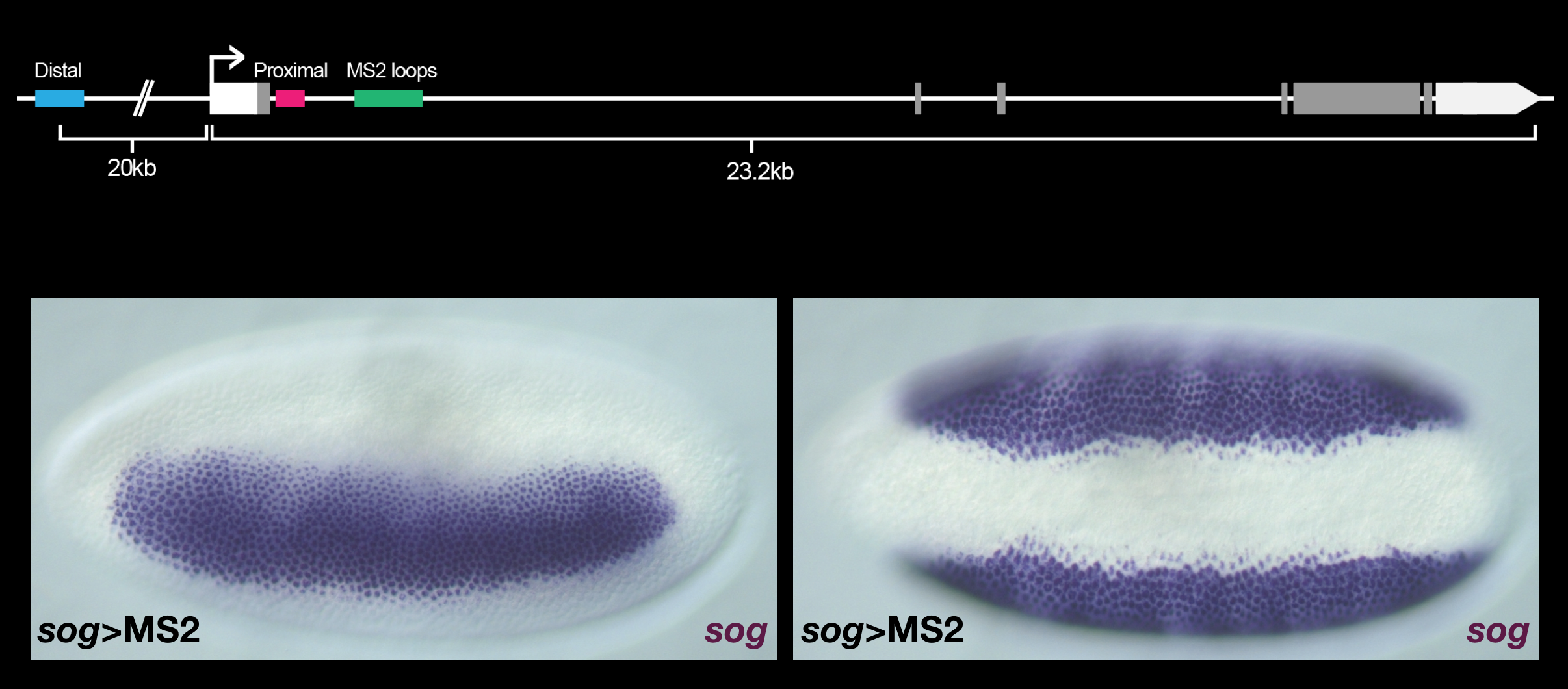
Conventional in situs demonstrate that the expression pattern of sog is unchanged, showing normal lateral (left) and ventral (right) expression boundaries.
CAGGTAG sites as proximal promoter elements
Some years ago we characterized the transcription factor that activates the early genes – Zelda (zinc-finger early Drosophila activator) (Liang et al., 2008). Zelda binds CAGGTAG sequencs, which are present directly upstream of the promoters of the early genes. These promoters also contain TATA boxes, and this organization of CAGGTAG and TATA sequences is conserved. We hypothesize that CAGGTAG act as proximal promoter sequences, interacting with TATA to ensure robust transcriptional activation. Preliminary results moving the CAGGTAG sites in the bnk proximal promoter 800 bp further upstream, shows weaker transcriptional output. We are currently quantifying these effects.
Function of Zelda’s unique ZF2 domain
Zelda contains six zinc fingers (see below), and we previously showed by gel shift DNA binding assays (Nien et al., 2011) and screens using PBMs (in collaboration with Tim Hughes at Univ of Toronto) that a recombinant Zelda protein comprising four fingers (ZF3-6) binds CAGGTAG sites, while an N-terminal protein comprising only the second zinc finger (ZF2) binds a G-rich motif.

We are currently investigating in vivo relevance for this motif by examining the expression of early-expressed genes in a ZF2 mutant background (Hamm et al., 2017). Once specific target genes are recovered, we will then search for G-rich sequences that might mediate the observed effects.